







Epithelioid soft tissue tumor with GLI-1 gene fusion or amplification is a recently described tumor entity (Note: tumors of uncertain differentiation/unknown origin classified as soft tissue tumors in the fifth edition of the WHO classification of head and neck tumors, 2022). Histologically, these tumors usually show multinodular or plexiform growth with round or ovoid tumor cells with hyaline or eosinophilic cytoplasm, often arranged in distinctive nest-like structures separated by an abundant network of slender dendritic vessels. IHC expression is nonspecific. A soft tissue tumor with altered GLI-1 gene is generally considered to be a low-grade sarcoma; however, the biological characteristics of this type of sarcoma is unclear based on the limited number of case reports. Interestingly, such tumors have a predilection for the head and neck region, mostly occurring on the tongue. As of article submission, only 11 cases of GLI-1-altered epithelioid soft tissue tumors have been reported in the English literature, none of which had recurrence or metastasis. To raise awareness of this solid tumor among oral pathologists and clinicians, we reported a case of epithelioid soft tissue tumor with GLI1 fusion that occurred on the tongue of a 56-year-old male with local lymph node metastasis and distant metastasis during follow-up. The accumulation of similar cases is essential to elucidate the biological behavior and to develop new therapeutic strategies.
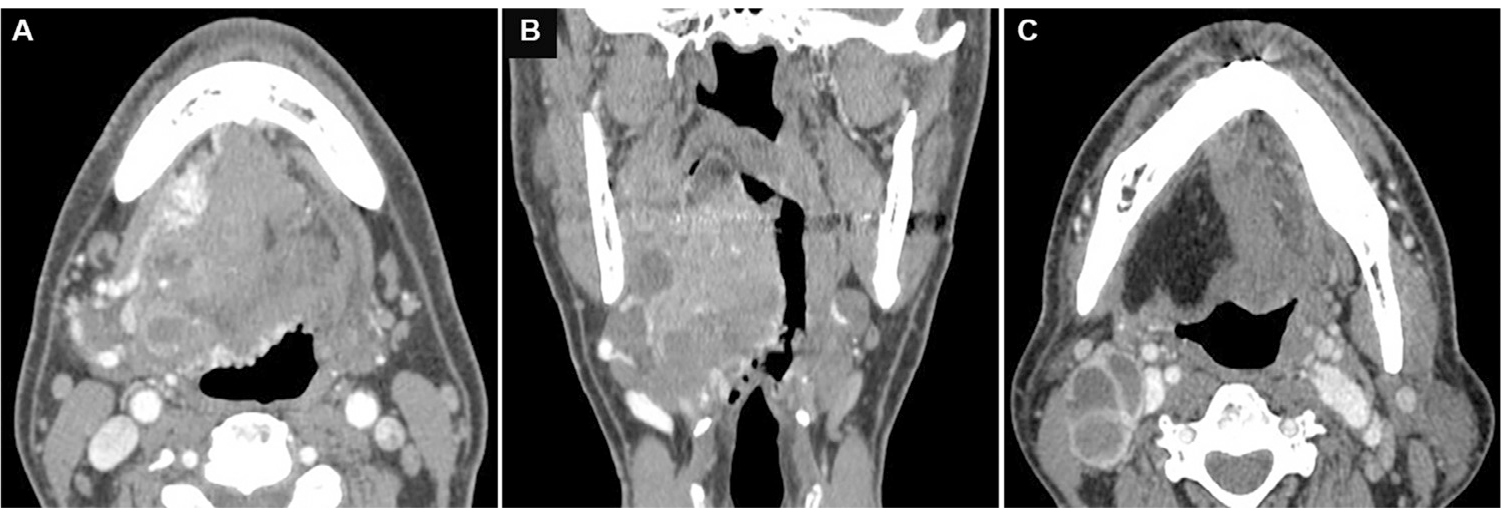
Case report: The patient, 56 years old, male, was admitted to the hospital in August 2015 due to speech disorders, swallowing difficulties and voice changes for 6 months. The patient had been seen in the ENT department of a local hospital where a large lump on the tongue was found. The patient had a heavy tobacco and alcohol addiction for 30 years and quit smoking and drinking 2 years ago. Medical history included hypertension and chronic bronchitis. Intraoral examination revealed a submucosal mass at the root of the right tongue extending into the right orofacial and lateral pharyngeal walls. Physical examination revealed a firm, mobile 2 cm right submandibular lymph node with mild pressing pain. Imaging: Imaging of the oral and maxillofacial region and neck revealed an ill-defined heterogeneous soft tissue tumor, size 6 cm × 5 cm × 3 cm, involving most of the tongue body, the right side of the tongue root, and the oropharynx.
Histology: histological examination of the biopsy showed a tumor under the surface mucosal epithelium with focal mucosal ulceration (A). In the central area, the tumor cells showed a lobular or plexiform growth pattern separated by a fibrotic mesenchyme with dilated blood vessels. There was an abundant, delicate, dendritic vascular network in the background of the tumor that divided the tumor cells into vague nest-like structures of varying sizes (B). In the peripheral areas, tumor cells grew in small nests or strips, or individually in a loosely adherent manner within the myxedematous interstitium. Mixed pseudoglandular structures were frequently present (C). Cytomorphologically, tumor cells were more uniform, round to epithelioid or short spindle-shaped, with slightly eosinophilic or hyaline cytoplasm (D). The nuclei were small or inconspicuous with mild to moderate nuclear pleomorphism. Nuclear division images were occasionally seen without areas of necrosis.
Immunohistochemistry (IHC): tumor cells were diffuse and strongly positive for the expression of CD56 (A) and cyclinD1 (B). (C) S100 was expressed in single cells or small clusters of cells, and most tumors were negative. (D) CD31 was negatively expressed in tumor cells.
In addition, membrane (but not nuclear) immunoreactivity for E-cadherin and β-catenin was observed in most cells. Perinuclear dot-like positivity was also observed for CD99. Tumor cells were negative for CK-pan, CK20, p63, EMA, S100, SMA, calponin, CD31, CD34, desmin, myogenin, MyoD1, CD45, CD20, CD3, CD56, CD57, NSE, chromogranin, synaptophysin, SOX10 and TFE3. Ki67 index was about 10%.
Diagnosis: Our oral pathologist initially diagnosed an unclassifiable malignant tumor; a major pathology consultation specialist initially diagnosed a low-grade malignant mesenchymal tumor (unclassifiable) with no clear IHC differentiation; another major pathology consultation specialist diagnosed a low-grade myoepithelial carcinoma.
The patient underwent an enlarged tumor resection and right suprahyoid neck dissection in September of that year.
Gross examination of the surgically excised sample revealed a 6 × 5 × 4 cm mass. The mass was tan to white in section and mostly solid, with hemorrhage in local areas (A). Microscopically, the morphology of the resected tumor was essentially similar to that seen in the previous biopsy. Under low magnification, the tumor was seen to have a multinodular or plexiform growth pattern with surrounding skeletal muscle and minor salivary gland infiltration (B). The surface squamous epithelium was focally ulcerated without abnormal hyperplasia. The tumor area consists of relatively homogeneous epithelioid and spindle cells arranged around many small thin-walled dendritic vessels. (C) Spindle cells and bundle arrangement were seen in local areas. (D) Tumor nests were seen locally protruding into the dilated vascular lumen.
Immunohistochemistry (IHC): tumor cells were negative for CKpan, CK7, CK5/6, CK14, CK18, p63, SMA, calponin, desmin, Melan-A, HMB45, and S100 with focal expression.
The biopsy and resected specimens were again sent to an external hospital for consultation by a soft tissue oncologist, who agreed with the diagnosis of low-grade myoepithelial carcinoma.
At 27 months after surgery, the patient had painless swelling of the right mandible for one month. Imaging revealed a peripherally enhanced, centrally necrotic enlarged lymph node with a diameter of 3.7 cm in the right submandibular region. Subsequently, further PET-CT revealed an enlarged lymph node of heterogeneous density in the right cervical II region and a mass in the soft tissue of the left sacrum with distending changes in the adjacent bone, suspicious of metastasis.
The patient was clinically diagnosed with myoepithelial carcinoma with suspicious lymph node and distant metastasis and then received treatment in a Class III-A cancer center. Puncture biopsy of the sacral mass confirmed a metastatic tumor with features similar to those of a tongue tumor. Afterwards, the patient underwent tumor resection and chemotherapy. The patient underwent cervical lymph node dissection (zones I-III) in January 2018, and an extended resection of the left sacral mass in May 2018. Both lymph node and sacral tumor were diagnosed as metastatic myoepithelial carcinoma by the institution. The patient remained under close monitoring and further follow-up 3 years after surgery for the sacral lesion, with no evidence of local recurrence or new metastases.
Recently, Dr. Yang Shaodong et al. reviewed the histological and immunological manifestations of this case and found similarities with the tumor reported by Xu et al.; FISH was immediately performed to detect the gene rearrangement status of GLI1 (12q13.3) and EWSR1 (22q12) (probes provided by Guangzhou LBP Medicine Science & Technology Co., Ltd. The results showed that 51.5% of the tumor cells had rearrangements in GLI1 gene, while no rearrangement was observed in EWSR1.
Based on morphologic and molecular genetic features, the case was finally diagnosed as an epithelioid soft tissue sarcoma with a GLI-1 gene fusion.
Details of this article can be found in: GLI1-altered epithelioid soft tissue tumor: A newly described entity with a predilection for the tongue. (Oral Surg Oral Med Oral Pathol Oral Radiol 2022;134:e14-e22) 10.1016/j.ooo.2021.10.007
A rare case jointly reported by LBP and Zhao Ming from Zhejiang Provincial People's Hospital is published 2022-08-30 11:34
Recently, a rare case reported by us in collaboration with Mr. Zhao Ming from Zhejiang Provincial People's Hospital was published in the International Journal of Gynecological Pathology: Uterine Tumor Resembling Ovarian Sex-Cord Stromal Tumor with Aggressive Histologic Features Harboring a GREB1-NCOA2 Fusion.
Case report: The patient, a 51-year-old female, previously healthy, went to hospital because of excessive menstruation and progressive dysmenorrhea for 6 months. A transvaginal pelvic ultrasound showed an enlarged uterus with a heterogeneous echogenicity of approximately 80×75×50 mm in the uterine cavity, which was considered to be a tumor of endometrial origin.
The mass tissue showed diffuse small blue round to epithelioid cells with active nuclear division images and focal tumor necrosis.
The tumor cells IHC expression: AE1/3, Cam5.2, ER and PR.
Initial diagnosis of suspected undifferentiated endometrial cancer;
The patient underwent panhysterectomy, bilateral salpingo‐oopherectomy and regional lymph node dissection. No other local or systemic treatment was received postoperatively, and there was no progression of disease at 12 months of follow-up.
Gross examination of the postoperative sample: the mass was a large, soft intrauterine polypoid mass with a maximum diameter of 8.5 cm and a grayish-yellow fleshy cut surface with hemorrhage, necrosis and cystic changes.
Morphology: under low magnification, tumor cells infiltrated the myometrium in cutting shape, similar to endometrial stromal sarcoma; tumor cells were mainly arranged in diffuse sheets and bundles, and multifocally in cords, the latter accounting for approximately 10% of the entire tumor. (A, B); some areas (5% of the tumor) had cystic changes and mulberry-like structures (C); the tumor cells had a homogeneous epithelioid and focal spindle-shaped morphology with mildly atypical vesicular nuclei, small and distinct nucleoli, and heterogeneous cytoplasmic eosinophilia (D, E); less than 5% of the areas contained rhabdomyoma cell populations (F); the histologic features included active nuclear division phases (up to 3/10 high power field ) (E), and with diffuse necrosis (G), as well as lymphovascular invasion (H).
The tumor stroma was sparse to focally sclerotic with an abundant vascular network. The uninvolved myometrium showed adenomyosis. The uterine cervix, ovary and fallopian tube had no abnormalities histologically. There were no tumors in the 26 pelvic cavities and periaortic lymph nodes.
IHC: diffuse positive for ER(A), PR, WT1, CD56, desmin(B), TLE-1 (﹥90% of cells); negative for Inhibin, melan-A, HMB45, epithelial membrane antigen, CD34, chromogranin A, PAX8, CD10, cyclinD1, SATB2, BCOR;
AE1/3 (20%–30% of cells. C), Cam5.2 (2%–5% of cells), calretinin (50%–60% of cells. D), CD99(70%–80% of cells. E), synaptophysin (30%–40% of cells), smooth muscle actin (SMA, 10%–20% of cells), h-caldesmon (40%–50% of cells)
Neither SMARCB1/INI-1 nor SMARCA4/BRG-1 was absent, and the Ki67 proliferation index was 20% (F).
Molecular assays:
RNA sequencing: in-frame fusion between exon 3 of GREB1 and exon 14 of NCOA2 (A).
FISH: Both GREB1 and NCOA2 were positive for rearrangement (B, C), which also confirmed the results of RNA sequencing.
Final diagnosis: uterine tumor resembling ovarian sex-cord tumor with GREB1-NCOA2 gene fusion.
In conclusion: Uterine tumor resembling ovarian sex-cord stromal tumor (UTROSCT) is a rare mesenchymal tumor with an uncertain direction of differentiation, mainly showing sec-cord differentiation with a wide range of histological manifestations and multi-phenotypic IHC features. Although it usually has a good prognosis, recurrence/metastasis may occur. Recently, several studies of UTROSCT have described novel fusion genes, including ESR1 and GREB1, as well as NCOA1-3. Genotypic and phenotypic correlations suggest that GREB1 rearranged tumors may be more aggressive.
For details, refer to: Uterine Tumor Resembling Ovarian Sex-Cord Stromal Tumor with Aggressive Histologic Features Harboring a GREB1-NCOA2 Fusion: Case Report with a Brief Review. International Journal of Gynecological Pathology. DOI: 10.1097/PGP.0000000000000849
The second case in the world: A rare case jointly reported by LBP and Zhao Ming from Zhejiang Provincial People's Hospital is published 2021-10-25 10:15
Recently, a rare case reported by us in collaboration with Zhao Ming from Zhejiang Provincial People's Hospital was published in the Journal of Cutaneous Pathology: BRAF-rearranged spindle cell mesenchymal neoplasm with a predominant lipofibromatosis-like neural tumor pattern and co-expression of CD34, S100 protein, and markers associated with perineurial differentiation. This very rare case, a subcutaneous tumor, is the second case reported in the literature and is easily misdiagnosed as another tumor.
Recently, a class of spindle cell mesenchymal neoplasm with a predominant lipofibromatosis-like neural tumor pattern and positive expression of S100 and/or CD34 were found to have unique molecular genetic features, often associated with abnormal translocations of various kinase genes, such as NTRK1-3, RAF1, RET, ROS1, ALK and MET. Among them, BRAF translocations are relatively rarer.
The authors report a case of a 54-year-old male patient with a 1.3-cm sized painless tumor mass located in the left dorsal subcutaneous tissue. The tumor tissue showed mild atypia with fascicular and spiral short spindle to ovoid cells mixed with subcutaneous adipose tissue and nerve bundles. A focal thick band of hyalinized stroma was seen. The tumor cells showed diffuse expression of S100 and CD34 and markers associated with perineurial differentiation in the multifocal areas, including EMA, GLUT1 and claudin-1. FISH results showed negative NTRK1, RET and ROS1 splitting, but positive BRAF splitting. Subsequently, the tumor was locally resected but recurred after 24 months.
It is reported that this is the second case of BRAF-rearranged spindle cell mesenchymal neoplasm with a predominant lipofibromatosis-like neural tumor pattern and expression of perineurial IHC markers, and with potential diagnostic pitfall, which could be misdiagnosed as a peripheral nerve sheath-associated tumor.
Multiple kinase-associated spindle cell mesenchymal tumors may exhibit histology of lipofibromatosis-like neural tumors. When the FISH test shows negative for NTRK gene rearrangement, an additional set of FISH-panel (including ALK, ROS1, RET, BRAF, etc.) can be considered to detect rare types of cases with kinase rearrangements, which provides a basis for targeted therapy for recurrent unresectable or metastatic tumors.
For details, refer to: BRAF-rearranged spindle cell mesenchymal neoplasm with a predominantlipofibromatosis-like neural tumor pattern and co-expression of CD34, S100protein, and markers associated with perineurial differentiation: A rare case with potential diagnostic pitfall. 2021. Journal of Cutaneous Pathology. DOI:10.1111/cup.14149








